Beta-Cell Dysfunction
Although abnormalities of insulin secretion in the pathophysiology of diabetes have often been neglected, they occur already early during the disease and can often already be demonstrated in subjects with normal glucose tolerance (first-degree relatives of type 2 diabetes).
Normal Insulin Secretion
After uptake of glucose pancreatic beta-cell glucose is rapidly degraded in oxidative glucose metabolism, leading to ATP formation. ATP is involved in beta-cell membrane depolarization.
The ADP/ATP ratio, the sulfonylurea receptor-1 (SUR 1) protein, which closes the adjacent potassium channel (potassium inward rectifier 6.2, KIR 6.2 channel). The closure of the potassium channels will decrease the membrane potential, which leads to opening of voltage-gated calcium channels; this induces the release of insulin-containing granules (Fig. 5). Upon stimulation with glucose, insulin is released with a short-lasting peak of a few minutes (so-called “first-phase”) followed by a slowly evolving second phase; the second phase lasts as long as the plasma glucose level is elevated.
Insulin Secretion in Type 2 Diabetes
In patients with type 2 diabetes, plasma glucose levels are elevated; and consequently, fasting plasma insulin. Although the insulin levels sometimes increase slightly after a meal in patients with type 2 diabetes mellitus this is considerably less than normal.
In studies in which glucose levels have been raised by glucose infusions (hyperglycemic clamps) to comparable levels in diabetic subjects and controls, it has become clear that second-phase insulin secretion is roughly 25% (IGT) to 50% decreased in type 2 diabetes. First-phase secretion is generally completely lost. In normoglycemic first-degree relatives insulin secretion is also diminished but to a lower extent, presumably on a genetic basis.
It is suspected that upon acquaintance of insulin resistance (obesity, physical inactivity) the pancreas that has already lower secretory capabilities can adapt less than normal, which might lead to decreased glucose tolerance or diabetes.
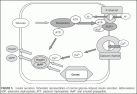 FIGURE 5 Insulin secretion. Schematic representation of normal glucose-induced insulin secretion.
FIGURE 5 Insulin secretion. Schematic representation of normal glucose-induced insulin secretion.
Abbreviations : ADP, adenosine dephosphate; ATP, adension triphosphate; IAAP, islet amyloid polypeptide.
It has been widely suggested that various mechanisms might further aggravate beta-cell insulin secretory dysfunction, among which glucose toxicity, lipotoxicity, and amyloid deposition.
Glucose Toxicity
Since over time insulin secretion appears to decrease in most patients, it has been proposed that glucose itself is toxic to beta cells.
This is in analogy with the situation in the “honeymoon period” in type 1 diabetes subjects: after the initial diagnosis of type 1 diabetes mellitus plasma glucose levels are lowered by injections with exogenous insulin. It has often been observed that dosages of exogenous insulin can be markedly decreased or even omitted during several months. It has been established that residual beta cells of these patients resume their insulin secretory function. Inevitably, in type 1 diabetes this is only a temporary improvement (due to the ongoing autoimmune destruction of remaining beta cells).
It may well be that in type 2 diabetes glucose is toxic as well. In pancreas beta cells oxidative glucose metabolism will also lead to formation of reactive oxygen species (ROS), which would damage beta cells (Fig. 6). Indeed, beta cells have low amounts of catalase and superoxide dismutase, proteins which normally metabolize the ROS. ROS can activate NF-kB activity, which would be proapoptotic.
Since it has been observed in an animal model of diabetes that pancreas duodenum homeobox-1 (PDX-1), a regulator of insulin gene transcription, is diminished by hyperglycemia, this could also be a mechanism of “glucose toxicity.”
Yet another mechanism may involve upregulation of uncoupling protein 2 (UCP-2) by high glucose that would lead to uncoupling of oxidative glucose metabolism from ATP formation in the mitochondrion leading to lower ATP.
Lipotoxicity
Although free fatty acids (FFA), also termed NEFA, acutely increase insulin secretion, chronic FFA overload diminishes beta-cell function. Type 2 diabetes subjects have often increased FFA due to insulin resistance to (adipocyte) lipolysis. It is now clear that high glucose inhibits beta-cell fatty acid oxidation, which may lead to accumulation of long-chain coenzyme A (LC-CoA).
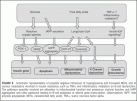 FIGURE 6 Schematic representation of possible negative influences of hyperglycemia and increased NEFA, and of various modulators involved in insulin resistance such as TNFa and inflammatory mediators on B-cell dysfunction.
FIGURE 6 Schematic representation of possible negative influences of hyperglycemia and increased NEFA, and of various modulators involved in insulin resistance such as TNFa and inflammatory mediators on B-cell dysfunction.
The pathways possibly involved are alteration in mitochondrial function and potassium channel function and IAPP aggregation and other pathways leading to B-cell apoptosis or altered gene transcription.
Abbreviations: IAPP, islet amyloid polypeptide; NEFA, nonesterified fatty acids; TNFa, tumor necrosis factor alpha.
This has been suggested to interfere with normal potassium channel activity, or to lead to activation of UCP-2, which would lead to uncoupling of oxidative glucose metabolism from ATP formation in the mitochondrion leading to lower ATP. FFA may diminish UCP-2 via augmentation of PPAR-gamma protein, which would lead to activation of UCP-2.
However, PPAR-gamma activation has numerous effects, and its overall importance in beta cells is therefore difficult to assess; for example, in animal models PPAR-gamma activation has been reported to enhance FFA oxidation in beta cells, which may in itself protect against lipotoxicity.
Yet another mechanism may involve synthesis of ceramide by FFA or generation of nitric oxide. In other tissues (muscle), degradation of ceramide has been shown to prevent FFA- induced insulin resistance almost completely; it is therefore conceivable that FFA act via ceramide formation in pancreas beta cells. Ceramide has been shown to inhibit insulin gene expression and has been implied in apoptosis via various pathways.
The importance of the insulin receptor signaling on insulin gene expression should not be underestimated, and may well harbor yet other mechanisms of lipotoxicity: via acyl-CoA FFA may inhibit insulin receptor signaling in beta cells via influences on IRS proteins, PI-3-kinase, or further downstream the insulin signaling cascade.
Islet Amyloid
It has been reported from postmortem studies in subjects with type 2 diabetes that most of the subjects’ pancreas islets contain amyloid in considerable quantities.
Amyloid consists of islet amyloid polypeptide (IAPP), or amylin, deposits. IAPP is normally contained in the insulin granule, and therefore cosecreted together with insulin (in a 10-fold lower quantity). Although amyloid is also present in islets of monkeys and cats, which have developed diabetes, it is absent in diabetic rodents, although rodents do secrete IAPP.
Small aggregates of IAPP are cytotoxic (in vitro), which has been suggested to be due to “channel formation” by aggregating IAPP molecules, which can lead to calcium influx into beta cells; another possibility is intracellular aggregation after interaction with liposomal membranes.
While hyperglycemia itself may accelerate IAPP aggregation, FFA (or NEFA) may enhance cytotoxicity of the aggregates. Although it is tempting to speculate that increased insulin secretion automatically leading to more IAPP secretion in insulin-resistant subjects would lead to IAPP aggregation, the finding that first-degree relatives secrete less IAPP (and insulin) than controls contradicts this hypothesis.
Since islet amyloid is absent in most insulin-resistant nondiabetic subjects, it seems more probable that amyloid formation is a relatively late occurrence during the pathophysiology of type 2 diabetes.
Michael Stumvoll
Department of Medicine, University of Leipzig, Leipzig, Germany
Barry J. Goldstein
Division of Endocrinology, Diabetes and Metabolic Diseases, Department of Medicine, Jefferson Medical
College of Thomas Jefferson University, Philadelphia, Pennsylvania, U.S.A.
Timon W. van Haeften
Department of Internal Medicine, University Medical Centre Utrecht, Utrecht, The Netherlands
REFERENCES