UCLA stem cell researchers move toward treatment for rare genetic nerve disease
Led by Dr. Peiyee Lee and Dr. Richard Gatti, researchers at the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA have used induced pluripotent stem (iPS) cells to advance disease-in-a-dish modeling of a rare genetic disorder, ataxia telangiectasia (A-T).
Their discovery shows the positive effects of drugs that may lead to effective new treatments for the neurodegenerative disease. iPS cells are made from patients’ skin cells, rather than from embryos, and they can become any type of cells, including brain cells, in the laboratory. The study appears online ahead of print in the journal Nature Communications.
People with A-T begin life with neurological deficits that become devastating through progressive loss of function in a part of the brain called the cerebellum, which leads to severe difficulty with movement and coordination. A-T patients also suffer frequent infections due to their weakened immune systems and have an increased risk for cancer. The disease is caused by lost function in a gene, ATM, that normally repairs damaged DNA in the cells and preserves normal function.
Developing a human neural cell model to understand A-T’s neurodegenerative process - and create a platform for testing new treatments — was critical because the disease presents differently in humans and laboratory animals. Scientists commonly use mouse models to study A-T, but mice with the disease do not experience the more debilitating effects that humans do. In mice with A-T, the cerebellum appears normal and they do not exhibit the obvious degeneration seen in the human brain.
Lee and colleagues used iPS cell–derived neural cells developed from skin cells of A-T patients with a specific type of genetic mutation to create a disease-in-a-dish model. In the laboratory, researchers were able to model the characteristics of A-T, such as the cell’s lack of ATM protein and its inability to repair DNA damage. The model also allowed the researchers to identify potential new therapeutic drugs, called small molecule read-through (SMRT) compounds, that increase ATM protein activity and improve the model cells’ ability to repair damaged DNA.
“A-T patients with no ATM activity have severe disease but patients with some ATM activity do much better,” Lee said. “This makes our discovery promising, because even a small increase in the ATM activity induced by the SMRT drug can potentially translate to positive effects for patients, slowing disease progression and hopefully improving their quality of life.”
What is ataxia-telangiectasia?
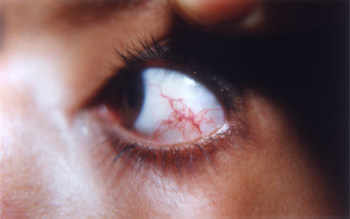
Ataxia-telangiectasia is a rare inherited disorder that affects the nervous system, immune system, and other body systems. This disorder is characterized by progressive difficulty with coordinating movements (ataxia) beginning in early childhood, usually before age 5. Affected children typically develop difficulty walking, problems with balance and hand coordination, involuntary jerking movements (chorea), muscle twitches (myoclonus), and disturbances in nerve function (neuropathy). The movement problems typically cause people to require wheelchair assistance by adolescence. People with this disorder also have slurred speech and trouble moving their eyes to look side-to-side (oculomotor apraxia). Small clusters of enlarged blood vessels called telangiectases, which occur in the eyes and on the surface of the skin, are also characteristic of this condition.
Affected individuals tend to have high amounts of a protein called alpha-fetoprotein (AFP) in their blood. The level of this protein is normally increased in the bloodstream of pregnant women, but it is unknown why individuals with ataxia-telangiectasia have elevated AFP or what effects it has in these individuals.
People with ataxia-telangiectasia often have a weakened immune system, and many develop chronic lung infections. They also have an increased risk of developing cancer, particularly cancer of blood-forming cells (leukemia) and cancer of immune system cells (lymphoma). Affected individuals are very sensitive to the effects of radiation exposure, including medical x-rays. The life expectancy of people with ataxia-telangiectasia varies greatly, but affected individuals typically live into early adulthood.
These studies suggest that SMRT compounds may have positive effects on all other cell types in the body, potentially improving A-T patients’ immune function and decreasing their susceptibility to cancer.
Additionally, the patient-specific iPS cell–derived neural cells in this study combined with the SMRT compounds can be an invaluable tool for understanding the development and progression of A-T. This iPS cell–neural cell A-T disease model also can be a platform to identify more potent SMRT drugs. The SMRT drugs identified using this model can potentially be applied to most other genetic diseases with the same type of mutations.
###
This research was supported by training and research grants from the California Institute of Regenerative Medicine, the National Institutes of Health, APRAT, A-T Ease and Scott Richards Foundation.
Syllaba and Henner first published descriptions of patients with ataxia-telangiectasia in 1926. They observed progressive choreoathetosis and ocular telangiectasia in 3 members of a single family. A gap of some 15 years occurred before the next report in 1941 by Louis-Bar, who described progressive cerebellar ataxia and cutaneous telangiectasia in a Belgian child. The syndrome subsequently received the name of Louis-Bar. Ataxia-telangiectasia was not described as a distinct clinical entity for another 16 years until Boder and Sedgwick in 1957 and Biemond in 1957, with the aid of autopsies, reported organ developmental abnormalities; neurologic manifestations; and a third major feature of the disease, recurrent sinopulmonary infection.
Ataxia-telangiectasia can best be classified, according to its major clinical and pathologic features, as a predominantly cerebellar form of spinocerebellar degeneration, which is transmitted as an autosomal recessive trait and evolves ultimately to include motor neuron disease, with spinal muscular atrophy and peripheral neuropathy.
Ataxia-telangiectasia can also be classified among the neurocutaneous syndromes, although not among the phakomatoses as originally proposed, because the vascular and cutaneous lesions of ataxia-telangiectasia are not congenital nevi but develop in the course of the disease as a progeric manifestation. Ataxia-telangiectasia should be considered among the immunodeficiency diseases, cancer-prone genetic disorders, chromosomal instability syndromes, disorders with abnormal radiosensitivity, syndromes with possible DNA-repair/processing defects, and (as is now evident) the progeroid syndromes.
The Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research: UCLA’s stem cell center was launched in 2005 with a UCLA commitment of $20 million over five years. A $20 million gift from the Eli and Edythe Broad Foundation in 2007 resulted in the renaming of the center. With more than 200 members, the Broad Stem Cell Research Center is committed to a multidisciplinary, integrated collaboration among scientific, academic and medical disciplines for the purpose of understanding adult and human embryonic stem cells. The center supports innovation, excellence and the highest ethical standards focused on stem cell research with the intent of facilitating basic scientific inquiry directed toward future clinical applications to treat disease. The center is a collaboration of the David Geffen School of Medicine at UCLA, UCLA’s Jonsson Cancer Center, the UCLA Henry Samueli School of Engineering and Applied Science and the UCLA College of Letters and Science.
###
Shaun Mason
.(JavaScript must be enabled to view this email address)
310-206-2805
University of California - Los Angeles