Linkage disequilibrium - The Genetics of Human Obesity
Linkage disequilibrium (Figure 2.2) is defined as the non-random association between two alleles at two different loci on the same chromosome. Although similar to linkage and based on the same fundamental genetic principles, the method is population rather than pedigree based and has the distinct advantage of being able to localize a linkage signal with far greater precision than is possible with traditional linkage alone (Jorde, 2000).
When a gene mutation first occurs in an individual, it forms part of a unique haplotype, which tends to dissipate over many successive generations as recombination events dislodge alleles at loci on the same chromosome (Figure 2.2). As with linkage, loci situated furthest away from the disease gene are most subject to separation from it during meiosis. After many generations (assuming non-lethality and lack of effect on reproductive potential) all that is left of the original chromosome in descendents is the disease gene flanked by a few alleles from the original haplotype which can be used to mark its position.
The ideal circumstances for fine LD mapping are encountered in founder communities (e.g. North-American Hutterites, Amish, Finnish or French-Canadian populations) because it is likely that the disease mutation was introduced by a single individual, producing the potential for a strong association between the disease and a specific haplotype in the current generation. Thus LD in such a population may be somewhat analogous to a family linkage study over many generations with an ancestral haplotype replacing a parental one. The ability to study many more ‘offspring’ (i.e. the present-day population) for linkage after many recombination events thus confers greater statistical power and much more precise localization than is possible with traditional linkage based methods.
The degree of LD depends on time elapsed since the mutation arose: in ‘older’ founder populations, the degree of LD often diminishes but due to more recombination events elsewhere, the ability to fine map a mutation may often be greater.
Important constraints of LD based methods include the fact that they depend on the disease of interest existing in suitable founder populations in a way not obscured by marker mutation within the population, new disease mutations, genetic drift, locus heterogeneity, population admixture or the existence of multiple disease susceptibility loci.
Problems related to population structure may also yield false positive results, that is, association without true linkage. Thus, assortive mating causing the eventual emergence of discrete genetic subpopulations (population stratification) and population admixture (where two genetically diverse populations have recently merged) may both lead to the association of unlinked loci.
Thus, additional studies are often required to determine whether a positive association is truly the result of genetic linkage between disease and marker loci.
The transmission disequilibrium test determines the transmission frequency of marker alleles from a heterozygous parent to an affected offspring. In the absence of true linkage, both alleles of a two allele marker locus will be transferred with equal frequency to an affected offspring. However, transmission of allele combinations with a frequency greater than that expected by chance is taken as evidence of true linkage.
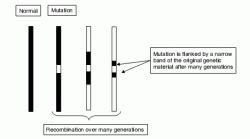
Figure 2.2 Linkage disequilibrium. A disease mutation initially arising on an ancestral haplotype becomes flanked by progressively smaller remnants of the original haplotype that are in LD with it and thus tend not to become separated from it by recombination events. These remnants may contain sequences which can be used to mark the position of the disease locus in a later generation. Although the detection of sequences derived from the ancestral haplotype may become more difficult after many generations, the precision of localization may be increased.
It is hoped that in the future it will be possible to perform whole genome LD scans but there are conceptual as well as practical difficulties to be overcome before this is likely to become a routine tool (Kruglyak, 1999).
Warden CH and Fisler JS
Katsanis N, Beales PL, Woods MO