Syndromic obesity - The Genetics of Human Obesity
Prader–Willi syndrome Long known to result from the deletion of a paternally inherited 4–4.5-Mb segment of chromosome 15, Prader–Willi syndrome
(PWS) exhibits the interesting phenomenon of ‘genetic imprinting’; when the deletion is inherited from the mother, Angelman syndrome (which does not have obesity as a cardinal feature) results but when the deletion is inherited from the father, loss of expression of one or more of the many C/D box, small nucleolar RNAs (snoRNAs) encoded within the paternally expressed small nuclear riboprotein N (SNRPN) locus permits the PWS phenotype to be expressed.
Figure 2.1 Protein products of genes known to be associated with human monogenic forms of obesity are shown in bold (see also Table 2.1). POMC, pro-opiomelanocortin; α-MSH, α-melanocyte stimulating hormone; PC-1, pro-hormone convertase-1; MC-1, 2 and 4, melanocortin-1, 2 and 4 receptors.
The ob gene mutation causing an absence of leptin is represented by (a) and leptin receptor mutations by (b). POMC mutations (c) disrupt cortisol biosynthesis and α-MSH-induced pigmentation as well as appetite regulation whereas PC-1 mutations (d) do not appear to have a major effect on pigmentation but instead lead to hypogonadism and glucoregulatory abnormalities. MC-4 mutations (e) are notable for causing little disturbance to regulatory pathways other than appetite and thus best represent common forms of non-syndromic obesity. 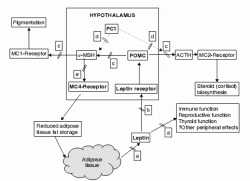
Generalized disruptions to the normal paternal expression of genes in this region (such as maternal uniparental disomy 15, imprinting centre mutations and specific balanced translocations with breakpoints within the paternally expressed SNRPN locus have also been reported to lead to PWS phenotype expression (Nicholls and Knepper, 2001). The precise cellular function of snoRNAs has yet to be determined but they are thought to play a part in the cellular localization and processing of RNA transcripts. To date, no link between common forms of human obesity and these abnormalities has been reported.
BBS
This syndrome, until recently considered to be autosomal recessive in nature and frequently confused with the Lawrence–Moon syndrome, is diagnosed on the basis of the presence of a constellation of major and minor features of which obesity (in 72–96 per cent of cases, preferentially distributed in the trunk and proximal limbs), retinal dystrophy, polydactyly, learning disabilities, male hypogonadism and renal abnormalities are most characteristic (Katsanis et al., 2001).
Table 2.1 Monogenic forms of human obesity
BBS prevalence varies widely, ranging from as little as 1:160 000 in ethnic Europeans to as much as 1:13 000 in populations such as Kuwait and Newfoundland where a founder effect has been postulated. Diagnostic difficulty has been compounded by a high degree of phenotypic variability, as much within as between families segregating the condition.
Since 1993, a combination of genome wide linkage scans in affected pedigrees and pooled sample homozygosity approaches have led to identification of the approximate position of six loci on chromosomes 16 (BBS2), 11 (BBS1), 3 (BBS3), 15 (BBS4), 2 (BBS5) and 20 (BBS6) with at least one further putative locus (BBS7) accounting for the 19–42 per cent of cases not explained by any of the above loci. BBS6 was first to be directly implicated in 2000 (Katsanis et al., 2000; Slavotinek et al., 2000) following its cloning as the gene responsible for a rather similar condition, the much rarer McKusick–Kaufman syndrome (MKKS). This knowledge, applied with increasingly detailed phenotyping, has permitted reclassification of some individuals previously thought to have MKKS as actually having BBS. Positional cloning of the BBS1, 2 and 4 loci has followed apace. Whereas BBS1 mutations appear sufficient for the development of classical BBS (Mykytyn et al., 2002), it has been suggested that three specific mutations at the other loci may be necessary for disease expression, at least in some cases (tri-allelic inheritance). Intense effort is currently underway to elucidate the precise function of these gene products in the hope that they may shed light on new mechanisms of body weight regulation.
New candidate genes may be identified by detailed ‘expression profiling’ whereby expression is found to be qualitatively (e.g. in appropriate tissues) or quantitatively (e.g. variable gene expression under different nutrient or energy balance conditions) consistent with a role in body weight regulation. Alternatively, loci which become implicated in obesity by the use of ‘positional’ genetic approaches may then be further investigated using the candidate gene approach.
Warden CH and Fisler JS
Katsanis N, Beales PL, Woods MO